class: inverse-intro .pull-left[ .my-title[<br>**riojaPlot**<br>] ## .my-black2[Stratigraphic diagrams in R] .footer1[Steve Juggins 2022] ] .pull-right[ 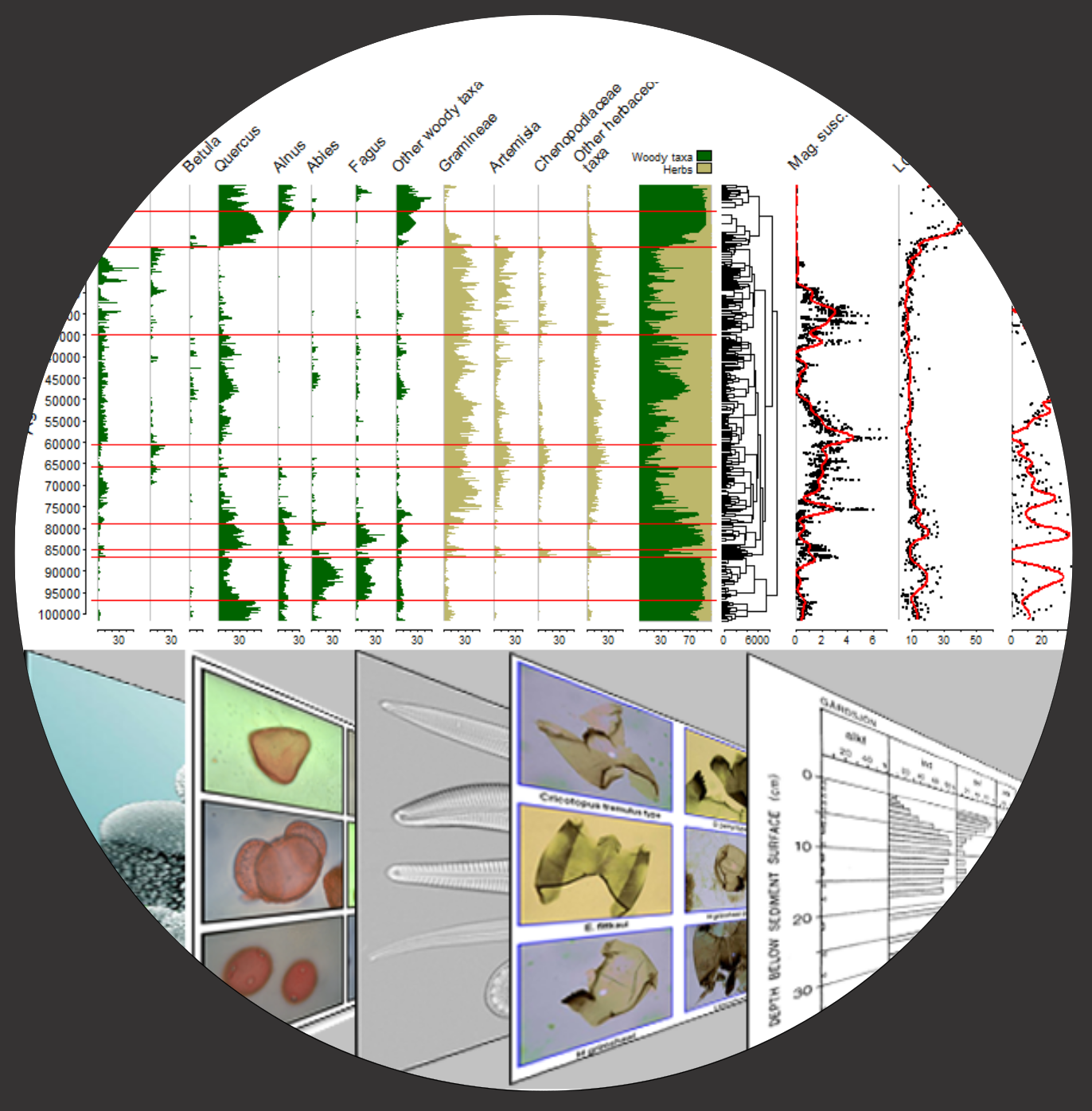 ] --- class: inverse-basic ## .footer-white[Getting started] .pull-right[ ] .pull-left[ ## .my-black3[Welcome to the riojaPlot <br><span style="color: #999999">gallery</span>] .intro-text[<br><br>.intro-code[**riojaPlot**] is an R package for producing publication quality stratigraphic diagrams. It's design is guided by four underlying principles illustrated in this gallery: - Easy to use - just call .intro-code[**riojaPlot**] with two data frames, the first containing variables to be plotted and the second containing one or more chronology variables for the y-axis, e.g. .intro-code[**riojaPlot(x, y)**]. See section .intro-high[[**Getting started**](#3)]. - Flexible - with a number of different plotting styles and ability to add a secondary y-axis, cumulative summary plot, zones and lithology. See section .intro-high[[**Pimp up my plot**](#14)]. - Build complex diagrams showing multiple datasets or proxies measured at different depth intervals, each with their own style. See section .intro-high[[**Plotting multiple datasets**](#28)]. - Customisable - .intro-code[**riojaPlot**] allows the use of user-defined plotting functions to customise plots and add additional features such as gam smoothes, breakpoints, or customised labels. See section .intro-high[[**Custom functions**](#33)]. Press .intro-high[**Control-F**] to search the gallery, press ".intro-high[**o**]" for an overview. .intro-code2[ Install with: install.packages("riojaPlot", repos="https://nsj3.r-universe.dev") or remotes::install_github('nsj3/riojaPlot', build_vignettes=TRUE, dependencies=TRUE) ]]] --- class: inverse-basic .pull-left[ ### Basic riojaPlot diagram ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name riojaPlot(poll, chron) ``` ] .pull-right-text[ ### .header-basic[Getting started]  .plot-text[This example shows the Abernethy Forest pollen dataset published by Birks and Mathews (1978) and included in package `rioja` (see `?aber` for details). The minimal call to `riojaPlot` passes two data frames or tibbles, the first containing variables to be plotted (the x-variables) and the second containing one or more variables to be used for the y-axis (chronlogy or y-variables). Unless otherwise specified, `riojaPlot` will use the first column in the chronology data and plot x-variables as lines and horizontal bars at sample positions, and show each column in equal-width plots. Missing values are omitted from each plot by default. Use argument `omitMissing=FALSE` to ratin missing values. This will leave gaps in line plots and lead to unexpected results with silhouettes. .reference-text[Birks, HH & Mathews, RW (1978). Studies in the vegetational history of Scotland V. Late Devensian and early Flandrian macrofossil stratigraphy at Abernethy Forest, Invernessshire. *New Phytologist* **80**, 455-84. ]]] --- class: inverse-basic .pull-left[ ### Basic diagram scaled for percentage data ```r # install.packages("riojaPlot", repos="https://nsj3.r-universe.dev") library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name riojaPlot(poll, chron, scale.percent=TRUE) ``` ] .pull-right-text[ ### .header-basic[Getting started]  .plot-text[Adding the argument `scale.percent=TRUE` scales each curve to equal data scaling and plots them with silhouettes, lines and horizontal lines (arguments `plot.poly=TRUE`, `plot.line=TRUE` and `plot.bar=TRUE`). ]] --- class: inverse-basic .pull-left[ ### Built-in plotting styles ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name # create logical vector of taxa > 5% max abundance max5 <- sapply(poll, max) > 5 poll5 <- poll[, max5] # remove rare taxa form data riojaPlot(poll5, chron, # default plot (silhouettes, lines and thin bars) scale.percent=TRUE) ``` ```r riojaPlot(poll5, chron, # silhouettes scale.percent=TRUE, plot.line=FALSE, plot.bar=FALSE) ``` ```r riojaPlot(poll5, chron, # lines scale.percent=TRUE, plot.line=TRUE, plot.poly=FALSE, plot.bar=FALSE) ``` ```r riojaPlot(poll5, chron, #bars scale.percent=TRUE, plot.line=FALSE, plot.poly=FALSE, plot.bar=TRUE, lwd.bar=2) ``` ] .pull-right-text[ ### .header-basic[Getting started]  .plot-text[Set arguments `plot.poly`, `plot.line` and `plot.bar` to control overall graph styles. Arguments may be give as a single logical value to turn on or off that style for the whole figure or a logical vector of `TRUE/FALSE` values to control the style of individual plots. Colours for each plot type are set with `col.bar`, `col.line` etc., line width (or bar thickness) with `lwd.bar`, `lwd.line` etc., and symbol type and size with `symb.cex` and `symb.pch`. ]] --- class: inverse-basic .pull-left[ ### Selecting columns to display ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name # select variables with maximum value > 5% max5 <- sapply(poll, max) > 5 max5_names <- names(max5[max5]) riojaPlot(poll, chron, selVars=max5_names, scale.percent=TRUE) ``` ] .pull-right-text[ ### .header-basic[Getting started]  .plot-text[To select which variables get plotted simply pass a character vector of column names to `selVars`. Variables will be plotted in the order they appear in this vector. ]] --- class: inverse-basic .pull-left[ ### Plotting groups I ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name # select variables with maximum value > 5% max5 <- sapply(poll, max) > 5 max5_names <- names(max5[max5]) types <- aber$names[, -1] riojaPlot(poll, chron, selVars=max5_names, groups=types, scale.percent=TRUE, plot.groups=TRUE) ``` ] .pull-right-text[ ### .header-basic[Getting started]  .plot-text[Individual columns can be assigned to a group and each group assigned a different colour. Colours can be changed using the argument `col.group=mycols` where `mycols` is a vector of 10 colour names or `rgb` values. ]] --- class: inverse-basic .pull-left[ ### Plotting groups II ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name # select variables with maximum value > 5% max5 <- sapply(poll, max) > 5 max5_names <- names(max5[max5]) types <- aber$names[, -1] riojaPlot(poll, chron, selVars=max5_names, groups=types, scale.percent=TRUE, plot.groups=TRUE, plot.cumul=TRUE) ``` ] .pull-right-text[ ### .header-basic[Getting started]  .plot-text[Cumulative plots can also be added showing the sum of each group. The names of the groups will be used in the cumulative plot legend so choose them wisely. ]] --- class: inverse-basic .pull-left[ ### Plotting groups III ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name # select variables with maximum value > 5% max5 <- sapply(poll, max) > 5 max5_names <- names(max5[max5]) types <- aber$names[, -1] types$Group <- factor(types$Group, levels=c("Trees", "Shrubs", "Herbs")) riojaPlot(poll, chron, selVars=max5_names, groups=types, scale.percent=TRUE, plot.groups=TRUE, plot.cumul=TRUE) ``` ] .pull-right-text[ ### .header-basic[Getting started]  .plot-text[The order of the groups and names in the legend is determined by the grouping variable. If this is a a character vector the groups will be ordered alphabetically. If it is a factor the groups will be ordered by the levels of the factor. To change the order of groups convert the group variable to a factor and adjust the order of the levels as in the above example. Change the relative width of the cumulative plot using `cumul.mult=N`, where N is scaling factor relative to the default width (ie. `cumul.mult=0.5` to make it half the default width. Cumulative plots are shown with filled polygons and no bounding lines. Use arguments `col.cumul.line` and `lwd.cumul.line` to show lines as well. ]] --- class: inverse-basic .pull-left[ ### Adding a zonation ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name # select variables with maximum value > 5% max5 <- sapply(poll, max) > 5 max5_names <- names(max5[max5]) types <- aber$names[, -1] types$Group <- factor(types$Group, levels=c("Trees", "Shrubs", "Herbs")) riojaPlot(poll, chron, selVars=max5_names, groups=types, scale.percent=TRUE, plot.groups=TRUE, plot.cumul=TRUE, clust.data.trans="sqrt", do.clust=TRUE, plot.clust=TRUE, plot.zones="auto") ``` ] .pull-right-text[ ### .header-basic[Getting started]  .plot-text[You can add zonation to the diagram in 3 ways. First, and easiest, to create a zonation based on the plotted data use arguments `do.clust=TRUE` to create the zonation and `plot.clust=TRUE` to add the dendrogram to the right-hand side of the diagram. Use argument `clust.data.trans="sqrt"` or `"scale"` to sqrt-transform the data or scale to zero man and unit variance before clustering (appropriate for percentage or geochemical data respectively, see `?chclust` for details). `riojaPlot` will use all variables in the zonation unless `clust.use.selected` is set to `TRUE`, in which case it will only use the displayed variables (if `selVars` is used to select which variables get plotted). Use argument `clust.width` to change width of the dendrogram as a fraction of the page width (default 0.05). With both these methods zone lines can be added using `plot.zones="auto"` to add significant zones, assessed using a broken stick model, or `plot.zones=N`, where N is the number of zones to plot. Zone colours and line thickness cn be changed with `col.zones` and `lwd.zones` respectively. The second way to add a diagram is to pre-calculate the zonation using function `chclust` and pass the chclust object to `riojaPlot` using the argument `clust`. Finally, the `chclust` object can added to the diagram as a separate component using function `addRPClust`. ]] --- class: inverse-basic .pull-left[ ### Adding exaggerations ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name # select variables with maximum value > 5% max5 <- sapply(poll, max) > 5 max5_names <- names(max5[max5]) riojaPlot(poll, chron, selVars=max5_names, scale.percent=TRUE, plot.exag=TRUE) ``` ] .pull-right-text[ ### .header-basic[Getting started]  .plot-text[Exaggerated curves can be added using argument `plot.exag=TRUE`. Use `exag.mult` to change the multiplication factor (default 2). Exaggeration colours are set with `col.exag`. Setting to `"auto"` will plot curves in transparent colour same as the main curve, with transparency set with `exag.alpha` (default 0.2). Passing a single TRUE or FALSE value controls plotting of all exaggerations curves. Pass a logical vector of TRUE / FALSE values to control plotting of exaggeration curves for each variable. Exaggerations are added as shaded silhouettes without bounding line to silhouette plots by default. Use arguments `col.exag.line` and `lwd.exag.line` and `col.exag=NA`, to show exaggerations as lines instead. ]] --- class: inverse-basic .pull-left[ ### Adding a secondary age or depth scale ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name # select variables with maximum value > 5% max5 <- sapply(poll, max) > 5 max5_names <- names(max5[max5]) riojaPlot(poll, chron, selVars=max5_names, yvar.name="Depth (cm)", sec.yvar.name="Age (years BP)", plot.sec.axis=TRUE, scale.percent=TRUE, plot.exag=TRUE) ``` ] .pull-right-text[ ### .header-basic[Getting started]  .plot-text[Use arguments `yvar.name` and `sec.yvar.name` to indicate which columns to use for the primary and secondary y-axes and set `plot.sec.axis=TRUE` to plot the axis. ]] --- class: inverse-basic .pull-left[ ### Saving a riojaPlot diagram ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name # select variables with maximum value > 5% max5 <- sapply(poll, max) > 5 max5_names <- names(max5[max5]) riojaPlot(poll, chron, selVars=max5_names, yvar.name="Depth (cm)", sec.yvar.name="Age (years BP)", plot.sec.axis=TRUE, scale.percent=TRUE, plot.exag=TRUE) ``` ] .pull-right-text[ ### .header-basic[Getting started] <img src="images/basic_plot_11.svg", width="60%")/> <img src="figures/basic_plot_11-1.svg", width="60%")/> .plot-text[`riojaPlot` figures can be saved to file or copied to the clipboard from R or RStudio. This will work but will not always give the best diagram because the plot is scaled to the window it is drawn in, and saving a plot that is already drawn to a different size or aspect ratio may result in unexpected gaps between axes or truncated text. To avoid this, place device calls around the plot to draw the figure to a graphics file. For a stand-alone figure `pdf` is appropriate. For a figure to include in a document or presentation `svg` will usually work best. This can be imported into recent versions of MS Office, or `png` files for earlier versions. Just be careful that if you get an error during the plot to remember to call `dev.off()` to return plotting to the screen. The above example shows 2 versions of the same diagram, both saved to svg (8 x 5 inches), the top saved directly from RStudio and the bottom by rendering directly to a file with `svg()`. ]] --- class: inverse-cosmetics ## .footer-white[Pimp up my plot] .pull-right[ ] --- class: inverse-cosmetics .pull-left[ ### Controlling y-axis ticks and limits ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name # select variables with maximum value > 5% max5 <- sapply(poll, max) > 5 max5_names <- names(max5[max5]) ytks1 <- seq(6000, 14500, by=500) riojaPlot(poll, chron, selVars=max5_names, yvar.name="Age (years BP)", sec.yvar.name="Depth (cm)", ymin=6250, ymax=14300, ytks1=ytks1, scale.percent=TRUE) ``` ] .pull-right-text[ ### .header-cosmetics[Pimp up my plot]  .plot-text[Use arguments `yvar.name` and `sec.yvar.name` to indicate which columns to use for the primary and secondary y-axis and set `plot.sec.axis=TRUE` to plot the axis. `riojaPlot` will try to find an appropriate range and tick values for the y-axis but it doesn't always get it right. To fine tune the y-axis, use arguments `ymin`, `ymax` and `yinterval` (and associated `sec.ymin` etc.) to fine tune the axis limits and intervals. `riojaPlot` will scale the y-axis exactly to the values provided. If these result in odd limits and interval values, supply a vector of tick values to argument `ytks1` and `ytks2` (for primary and secondary axes respectively). y-axis tick values can be rotated using `las.yaxis`. In the example above samples range in age from 6295 to 14229 years BP, so we set `ymin=6250` and `ymax=14300` and define `ykts1` to run from 6000 to 14500 at 500 year intervals. If the y-axis variable is a character vector the axis will be scaled from 1 to the number of samples and the sample labels will be used as tick values. ]] --- class: inverse-cosmetics .pull-left[ ### Controlling x-axis ticks and limits ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name # select variables with maximum value > 5% max5 <- sapply(poll, max) > 5 max5_names <- names(max5[max5]) ytks1 <- seq(6000, 14500, by=500) riojaPlot(poll, chron, selVars=max5_names, yvar.name="Age (years BP)", sec.yvar.name="Depth (cm)", ymin=6250, ymax=14300, ytks1=ytks1, plot.sec.axis=TRUE, scale.percent=TRUE, tcl=0.2, plot.top.axis=TRUE) ``` ] .pull-right-text[ ### .header-cosmetics[Pimp up my plot]  .plot-text[For percentage data `riojaPlot` will print ticks at intervals determined by `x.pc.inc` (default 10) and omit the initial zero (`x.pc.omit0=TRUE`). The minimum width of a individual plot is controlled by `min.width.pc` (default 10). For non-percentage data `riojaPlot` will scale the axis from minimum to maximum value either try to find sensible tick values or just show the minimum and maximum of the axis to avoid label crowding (`scale.minmax=TRUE`). To have fine-control over the x-axis range in non-percentage diagrams supply a data frame with two columns containing the mimimum and maximum values for each variable to argument `minmax`. x-axis tick values can be rotated using `las.xaxis=2` and tick lengths can be adjusted with `tcl` (default -0.2, use positive values to place ticks inside the plot). Set argument `plot.top.axis` and `plot.bottom.axis` to `TRUE`/`FALSE` to plot top and bottom x-axes respectively. A vertical baseline is added to each plot (at zero for percentage scaling or minimum value for normal scaling). The colour and thickness of this line change be changed with `col.baseline` and `lwd.baseline`. Set `col.baseline=NA` to omit this line. ]] --- class: inverse-cosmetics .pull-left[ ### Controlling x- and y-axis labels ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names # replace column names will full taxon names colnames(poll) <- names$Name # select variables with maximum value > 5% max5 <- sapply(poll, max) > 5 max5_names <- names(max5[max5]) ylab <- expression(Age~"("^{14}*C~years~BP*")") riojaPlot(poll, chron, selVars=max5_names, yvar.name="Age (14C years BP)", ylabel=ylab, scale.percent=TRUE, labels.break.n=15, labels.italicise=TRUE, srt.xlabel=45) ``` ] .pull-right-text[ ### .header-cosmetics[Pimp up my plot]  .plot-text[`riojaPlot` will label the x- and y-axes with the columns names of the corresponding data frames. Names can be changed by either changing the names of the data frame or by passing a character vectors with custom names. For the primary and secondary y-axes use arguments `ylabel` and `sec.ylabel`. See next page for customising the x-labels. If there is a single y-axis the label will be placed on the left of the diagram, or at the top if a secondary y-axis is plotted. With a single y-axis, use argument `ylabPos` to tweak the position to the label if `riojaPlot` gets it wrong. Labels for the x-variables and chronology can be rotated using `srt.xlabel` `srt.ylabel` and the space between the label and plot tweaked with `xlabPos`. Use `centre.xlabel` to centre the label when plotted horizontally (`srt.xlabel=0`). Long names can be split into two or more lines using `labels.break.long=TRUE` (the default) amd `labels.break.n` used to set the maximum number of characters per line (default 20). Names can be italicised using `labels.italicise=TRUE`. Mathematical `expressions` can be used for labels - the above example plots the data on a radiocarbon timescale and uses an expression to produce an appropriate label (see `?mathplot`). The size of text for various plotting elements can be changed with the following arguments: axis values: `cex.xaxis`, `cex.yaxis`; axis labels: `cex.ylabel`, `cex.xlabel`; zone names: `cex.zones`; groups names: `cex.cumul`. ]] --- class: inverse-cosmetics .pull-left[ ### Custom x-axis labels ```r library(dplyr) fpath <- system.file("extdata/maule2020geochem.txt", package="riojaPlot") maule <- readr::read_delim(fpath, skip=158, show_col_types=FALSE) maule.data <- maule %>% select(-(1:4)) maule.chron <- maule %>% select(1:4) colnames(maule.data) ``` ``` ## [1] "MS" "TS" "TC" "TIC" "TOC" "BioSi" "TN" "d15N" ## [9] "d13C" ``` ```r xnames <- c("Mag. Susc.", "Total sulphur", "Total carbon", "Total inorg. carbon", "Total org. carbon", "Biogenic silica", "Total Nitrogen", "d15N", "d13C") xnames[8] <- expression(delta^15*N) xnames[9] <- expression(delta^13*C) riojaPlot(maule.data, maule.chron, groups=groups, yvar.name="age_BP", xlabels=xnames, ylabel="Age (years BP)", plot.bar=FALSE, plot.poly=FALSE, plot.line=TRUE, col.line="grey90", plot.symb=TRUE, symb.cex=0.3) ``` ] .pull-right-text[ ### .header-cosmetics[Pimp up my plot]  .plot-text[The names of the x-variables can be customised by passing a vector of new names to `xlabels`. These names can contain R `expressions`. In this example we use geochemical data from Laguna del Maule published by Frugone-Álvarez et al. (2020) and downloaded from the NOAA Paleoclimate data archive (https://www.ncei.noaa.gov/products/paleoclimatology). The data we plot here contain 9 geochemical variables. In the above example we replace the abbreviated names of the column with full variable names and use expressions to label the delta 15N / 13C variables. Note that long names using expressions cannot be broken or split into several lines (though see `?atop` for creating multi-line expressions). .reference-text[Frugone-Álvarez, M. et al. (2020) Volcanism and climate change as drivers in Holocene depositional dynamic of Laguna del Maule (Andes of central Chile - 36S). Climate of the Past, DOI 10.5194/cp-2019-147. ]]] --- class: inverse-cosmetics .pull-left[ ### Page layout and margins ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages colnames(poll) <- substring(colnames(poll), 1, 4) # select variables with maximum value > 5% max5 <- sapply(poll, max) > 10 max5_names <- names(max5[max5]) riojaPlot(poll, chron, selVars=max5_names, # bottom plot scale.percent=TRUE, labels.break.n=15, labels.italicise=TRUE, srt.xlabel=0, yBottom=0.1, yTop=0.45, cex.xlabel=0.6, xSpace=0.02, plot.poly=FALSE) riojaPlot(poll, chron, selVars=max5_names, # top plot scale.percent=TRUE, labels.break.n=15, labels.italicise=TRUE, srt.xlabel=0, yBottom=0.55, yTop=0.9, cex.xlabel=0.6, xSpace=0.02, start.new.plot=FALSE) ``` ] .pull-right-text[ ### .header-cosmetics[Pimp up my plot]  .plot-text[ `riojaPlot` will scale the diagram to fit the plotting device (ie. screen or file if plotting to pdf, svg etc.), taking into account the size of any labels. Sometimes it may get the scaling wrong and labels are truncated. You may also want to fix the fix of the diagram so multiple diagrams are exactly the same height. In these cases the margins around the plotting area can be changed using arguments `xLeft`, `xRight`, `yBottom` and `yTop`. These arguments take values as fraction of the page width / height. The space between individual plots can be changed using `xSpace`. The plotting of the y-axis can be suppressed by setting `plot.yaxis=FALSE`, and a second plot can be added to the page by setting `start.plot.new=FALSE`; ]] --- class: inverse-cosmetics .pull-left[ ### Changing colours ```r library(dplyr) fpath <- system.file("extdata/maule2020geochem.txt", package="riojaPlot") maule <- readr::read_delim(fpath, skip=158, show_col_types = FALSE) maule.data <- maule %>% select(-(1:4)) maule.chron <- maule %>% select(1:4) # create logical vectors to control plotting of lines, # symbols etc for each variable selsymb <- rep(FALSE, 9) selsymb[c(1, 8:9)] <- TRUE selbar <- rep(FALSE, 9) selline <- rep(TRUE, 9) selline[1] <- FALSE cols <- c("black", rep("orange4",4), rep("steelblue3", 2), rep("tan2", 2)) riojaPlot(maule.data, maule.chron, yvar.name="age_BP", ymin=-100, ymax=13300, yinterval=500, plot.bar=selbar, plot.poly=FALSE, plot.line=selline, col.line=cols, plot.symb=selsymb, col.symb=cols, plot.groups=TRUE, symb.cex=0.5) ``` ] .pull-right-text[ ### .header-cosmetics[Pimp up my plot]  .plot-text[ The colours for bars, lines, silhouettes and symbols can also be set for all plots by passing a single colour to `col.line` etc., or set for each plot individually by passing a vector of colours. Similarly, the plot type can be set for each plot by passing a logical vector to `plot.bar` etc. The colours for individual plots can can also be changed by grouping variables using the argument `groups` (see above in "Getting started"). This mechanism is more useful to, for example, plot taxa from different ecological groups in different colours. Passing a vector of colours is more useful for setting ad-hoc colours to different variables. ]] --- class: inverse-cosmetics .pull-left[ ### Changing plot widths (percentage scaling) ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages # replace column names will full taxon names colnames(poll) <- names$Name mx <- sapply(poll, max) # numeric vector of widths, set to 1 widths <- rep(1, ncol(poll)) # generate numeric vector of x-intervals, set to 10 inc <- rep(10, ncol(poll)) # generate character vector of column names > 2% selTaxa <- names(mx[mx > 2]) # generate a logical vector of columns > 5% sel <- which(mx < 5) widths[sel] <- 5 # set widths to 5 times normal width inc[sel] <- 1 # set x-interval to 1% riojaPlot(poll, chron, selVars=selTaxa, scale.percent=TRUE, graph.widths=widths, min.width.pc=5, x.pc.inc=inc, cex.xaxis=0.5, srt.xlabel=45, cex.xlabel=0.7, las.xaxis=2) ``` ] .pull-right-text[ ### .header-cosmetics[Pimp up my plot]  .plot-text[ For percentage diagrams the width of each plot is scaled uniformly so so unit percentage width is the same for all variables. We can change these using the style `graph.widths` which takes a vector of relative widths. We can also change the x-axis tick-interval for each graph by supplying a numeric vector to style `x.inc.pc`. In the above example we expand the scale of rare taxa. After hiding columns with maximum relative abundance < 2%, we expand those taxa with max abundance < 5% by plotting with 5 * normal width, and set the x-interval for these to 1% (rather than the default of 10%). ]] --- class: inverse-cosmetics .pull-left[ ### Changing plot widths (normal scaling) ```r library(dplyr) fpath <- system.file("extdata/maule2020geochem.txt", package="riojaPlot") maule <- readr::read_delim(fpath, skip=158, show_col_types = FALSE) maule.data <- maule %>% select(-(1:4)) maule.chron <- maule %>% select(1:4) # create logical vectors to control plotting of lines, # symbols etc for each variable selsymb <- rep(FALSE, 9) selsymb[c(1, 8:9)] <- TRUE selbar <- rep(FALSE, 9) selline <- rep(TRUE, 9) selline[1] <- FALSE cols <- c("black", rep("orange4",4), rep("steelblue3", 2), rep("tan2", 2)) widths <- rep(1, ncol(maule.data)) widths[8:9] <- 2 riojaPlot(maule.data, maule.chron, yvar.name="age_BP", ymin=-100, ymax=13300, yinterval=500, plot.bar=selbar, plot.poly=FALSE, plot.line=selline, col.line=cols, plot.symb=selsymb, col.symb=cols, plot.groups=TRUE, symb.cex=0.5, graph.widths=widths) ``` ] .pull-right-text[ ### .header-cosmetics[Pimp up my plot]  .plot-text[ In this example we repeat the diagram from Laguna del Maule but make the delta 15N and 13C columns twice the width of the other variables. ]] --- class: inverse-cosmetics .pull-left[ ### Changing the order of variables I ```r library(riojaPlot) library(rioja) rlgh.names <- RLGH$names rlgh.diat <- RLGH$spec rlgh.mx <- sapply(rlgh.diat, max) # plot only common taxa rlgh.sel <- names(rlgh.mx[rlgh.mx > 2]) rlgh.chron <- RLGH$depths # data has 210Pb age based on years before coring (1980) # Add new column with Years (CE) rlgh.chron$Year <- 1980 - rlgh.chron$Age riojaPlot(rlgh.diat, rlgh.chron, yvar.name="Year", scale.percent=TRUE, y.rev=FALSE, selVars=rlgh.sel, ymax=1980, yinterval=10, xlabels=RLGH$names$TaxonName, cex.xlabel=0.7, labels.break.n=25, wa.order="bottomleft") ``` ] .pull-right-text[ ### .header-cosmetics[Pimp up my plot]  .plot-text[ There are several ways to reorder the variables in a `riojaPlot`: (1) reorder the variables in the original data frame, (2) create a character vectors of columns names in the required order and pass to `selVars`, and (3) reorder based on stratigraphic trends. The example above using diatom data from the Round Loch of Glenhead, an acidified loch in SW Scotland (see ?RLGH for details and citation) illustrates this last approach. We use argument `wa.order="bottomleft"` to order taxa from those present at the base of the core on the left to those at the top on the right. ]] --- class: inverse-cosmetics .pull-left[ ### Changing the order of variables II ```r library(riojaPlot) library(rioja) library(dplyr) rlgh.names <- RLGH$names rlgh.diat <- RLGH$spec rlgh.mx <- sapply(rlgh.diat, max) # plot only common taxa rlgh.sel <- names(rlgh.mx[rlgh.mx > 3]) rlgh.chron <- RLGH$depths rlgh.chron$Year <- 1980 - rlgh.chron$Age optima <- WA(SWAP$spec, SWAP$pH)$coefficients %>% data.frame() %>% tibble::rownames_to_column(var="CODE") optima2 <- data.frame(CODE=colnames(rlgh.diat)) %>% dplyr::left_join(optima, by="CODE") %>% mutate(Group=case_when(Optima <= 5.1 ~ "Acidobiont.", Optima > 5.1 & Optima < 5.4 ~ "Acidophil.", TRUE ~ "Acid intol."), Group=factor(Group, levels=c("Acid intol.", "Acidophil.", "Acidobiont."))) %>% mutate(Optima=ifelse(is.na(Optima), 5.5, Optima)) %>% arrange(desc(Optima)) %>% select(CODE, Group, Optima) rlgh.sel <- optima2 %>% filter(CODE %in% rlgh.sel) %>% pull(CODE) rlgh.names <- RLGH$names %>% left_join(optima2, by="CODE") %>% mutate(name2=paste0(TaxonName, " (", round(Optima, 1), ")")) riojaPlot(rlgh.diat, rlgh.chron, groups=optima2, selVars=rlgh.sel, yvar.name="Year", scale.percent=TRUE, y.rev=FALSE, ymax=1980, yinterval=10, xlabels=rlgh.names$name2, labels.italicise=TRUE, cex.xlabel=0.7, plot.poly=FALSE, plot.line=FALSE, lwd.bar=3, plot.groups=TRUE, plot.cumul=TRUE, x.pc.inc=5, labels.break.n=30, col.group=c("mediumblue", "darkgrey", "red2"), cumul.mult=0.2) ``` ] .pull-right-text[ ### .header-cosmetics[Pimp up my plot]  .plot-text[To have complete control over the order of taxa in a diagram we need to create a character vector with taxa in the required order. In this example we repeat the diagram from the Round Loch of Glenhead but this time sort the taxa according to their pH optima. We also group the taxa into three pH classes based on their pH optima and display the groups and cumulative plot, and append the pH optimum to the name of each taxon. ]] --- class: inverse-cosmetics .pull-left[ ### Grouping samples ```r library(riojaPlot) library(rioja) data(Ponds) # reorder the rows in decreasing TP o <- order(Ponds$env$TP, decreasing=TRUE) ponds.diat <- Ponds$spec[o, ] ponds.TP <- data.frame(TP=round(Ponds$env$TP[o]), LakeName=Ponds$env$Name[o]) # replace taxon codes with names colnames(ponds.diat) <- Ponds$names$Name # remove rare species mx <- apply(ponds.diat, 2, max) ponds.sel <- colnames(ponds.diat)[mx > 10] ponds.TP <- ponds.TP %>% mutate(bar.cols=case_when(TP < 100 ~ "green", TP >= 100 & TP < 200 ~ "blue", TP >= 200 & TP < 500 ~ "red", TRUE ~ "orange")) ylab <- expression(Total~Phosph.~(mu*gL^{-1})) riojaPlot(ponds.diat, ponds.TP, selVars=ponds.sel, yvar.name="LakeName", sec.yvar.name="TP", sec.ylabel=ylab, sec.yinterval = 20, plot.sec.axis=TRUE, scale.percent=TRUE, plot.poly=FALSE, plot.line=FALSE, plot.bar=TRUE, sep.bar=TRUE, col.sep.bar=ponds.TP$bar.cols, lwd.bar = 10, cex.xlabel=0.5, cex.yaxis=0.6, cex.xaxis=0.5, wa.order="bottomleft", labels.italicise=TRUE, las.xaxis=2) ``` ] .pull-right-text[ ### .header-cosmetics[Pimp up my plot]  .plot-text[ It is currently only possible to plot different samples with different coloured bars. This example plots surface sediment diatom data from small ponds and pools in SE England collected by Bennion (1994) (see ?rioja::Ponds) for details. Here we arrange the samples along the TP gradient and plot the data with Lake name and total phosphorus (TP) as a secondary y-axis. We also classify the lakes into different groups based on their TP, then plot these different samples with different colours. We do this by creating a character vector of colour names, that corresponds to the samples, then pass this to style `col.sep.bar` and set `sep.bar=TRUE`. We also create an expression to label the TP axis in the correct units. ]] --- class: inverse-cosmetics .pull-left[ ### User define zones ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages colnames(poll) <- names$Name mx <- sapply(poll, max) selTaxa <- names(mx[mx > 2]) rp <- riojaPlot(poll, chron, selVars=selTaxa, yvar.name="Age (years BP)", ymin=6000, ymax=14300, yinterval=500, scale.percent=TRUE, cex.xaxis=0.5, cex.xlabel=0.7, las.xaxis=2) # define zone lines at 11000 and 13000 years BP myzones <- c(11000, 13000) addRPZone(rp, myzones, col="red") # add shaded zone from 6000 to 8000 years BP addRPZone(rp, 6000, 8000) # change colour and shading addRPZone(rp, 13000, 13500, col="blue", alpha = 0.05) ``` ] .pull-right-text[ ### .header-cosmetics[Pimp up my plot]  .plot-text[Function `addRPZone` can be used to add horizontal zone lines or shaded rectangles to an existing plot. The zones above are plotted over the existing diagram so if using a solid fill set alpha to a low number to produce a transparent colour. It is possible to plot zones or other annotations behind by defining a custom function (see section on "Custom functions"). ]] --- class: inverse-cosmetics .pull-left[ ### Column with zone names ```r library(rioja) library(riojaPlot) library(dplyr) poll <- aber$spec chron <- aber$ages colnames(poll) <- aber$names$Name mx <- sapply(poll, max) selTaxa <- names(mx[mx > 2]) clust <- chclust(dist(sqrt(poll))) chron$Zone <- cutree(clust, k=5) zones <- chron %>% group_by(Zone) %>% summarise(zm=mean(`Age (years BP)`)) %>% mutate(name=paste("Zone", Zone)) %>% select(-Zone) zone.names <- paste("Zone", 1:5) riojaPlot(poll, chron, selVars=selTaxa, yvar.name="Age (years BP)", ymin=6000, ymax=14300, yinterval=500, scale.percent=TRUE, cex.xaxis=0.5, cex.xlabel=0.7, xRight=0.82) |> addRPZoneNames(zones, xRight=0.9, cex=0.6) |> addRPClustZone(clust, col="red") |> addRPClust(clust) ``` ] .pull-right-text[ ### .header-cosmetics[Pimp up my plot]  .plot-text[This example shows how to add a column with zone names using function `addRPZoneNames`. We first cluster the data with `chclust`, then extract the zone membership from the `chclust` object, calculate the mean age of each zone and combine into a data frame with zone names. We then construct a composite diagram by chaining together the pollen plot, zones and cluster dendrogram and finally add zone lines. In this example we use the pipe to chain the 4 function calls together. ]] --- class: inverse-multi ## .footer-white[Plotting multiple datasets] .pull-right[ ] --- class: inverse-multi .pull-left[ ### Plotting a second dataset ```r library(riojaPlot) library(rioja) library(dplyr) rlgh.names <- RLGH$names rlgh.diat <- RLGH$spec rlgh.mx <- sapply(rlgh.diat, max) # plot only common taxa rlgh.sel <- names(rlgh.mx[rlgh.mx > 3]) rlgh.chron <- RLGH$depths rlgh.chron$Year <- 1980 - rlgh.chron$Age SWAP.wa <- WA(SWAP$spec, SWAP$pH) rlgh.pH <- predict(SWAP.wa, RLGH$spec) # convert to data frame with single column rlgh.pH <- data.frame(`DI-pH (SWAP)`=rlgh.pH$fit[, 1], check.names=FALSE) rp <- riojaPlot(rlgh.diat, rlgh.chron, yvar.name="Year", scale.percent=TRUE, y.rev=FALSE, selVars=rlgh.sel, ymax=1980, xlabels=RLGH$names$TaxonName, cex.xlabel=0.7, labels.break.n=25, xRight=0.9) riojaPlot(rlgh.pH, rlgh.chron, riojaPlot=rp, yvar.name="Year", scale.minmax=FALSE, plot.bar=FALSE, plot.symb=TRUE, symb.cex=0.6) ``` ] .pull-right-text[ ### .header-multi[Plotting multiple datasets]  .plot-text[To combine different datasets we plot the first with function `riojaPlot` and save the reuslting `riojaPlot object`. Additional datasets are then added by passing this object to further calls to `riojaPlot`. Each call to `riojaPlot` must have it's own x- and y- data frames and will inherit axis scaling and font sizes from the initial call but these can be overridden. Function `riojaPlot2` is a wrapper around `riojaPlot` that takes a riojaPlot object as the first argument so it can be used with a pipe (`%>%` or `|>`). When combining multiple datasets we just need to specify the right-hand position of each plot in fractions of the page width. In the example above we plot the data from the Round Loch of Glenhead and add a pH reconstruction to the right of the figure. Use argument `xGap` to change the gap between datasets (as fraction of the page width). ]] --- class: inverse-multi .pull-left[ ### Adding a zonation with multiple datasets ```r library(riojaPlot) library(rioja) library(dplyr) rlgh.names <- RLGH$names; rlgh.diat <- RLGH$spec rlgh.mx <- sapply(rlgh.diat, max); rlgh.sel <- names(rlgh.mx[rlgh.mx > 3]) rlgh.chron <- RLGH$depths; rlgh.chron$Year <- 1980-rlgh.chron$Age SWAP.wa <- WA(SWAP$spec, SWAP$pH); rlgh.pH <- predict(SWAP.wa, RLGH$spec) rlgh.pH <- data.frame(`DI-pH (SWAP)`=rlgh.pH$fit[, 1], check.names=FALSE) clust <- chclust(dist(sqrt(rlgh.diat))) rp1 <- riojaPlot(rlgh.diat, rlgh.chron, yvar.name="Year", scale.percent=TRUE, y.rev=FALSE, selVars=rlgh.sel, ymax=1980, xlabels=RLGH$names$TaxonName, cex.xlabel=0.7, labels.break.n=25, x.pc.inc=5, xRight=0.82) rp2 <- riojaPlot(rlgh.pH, rlgh.chron, riojaPlot=rp1, yvar.name="Year", scale.minmax=FALSE, plot.bar=FALSE, plot.symb=TRUE, symb.cex=0.6, xRight=0.92) rp3 <- addRPClustZone(rp2, clust, nZone=3, xRight=0.92, col="red") addRPClust(rp3, clust, xLeft=0.93) ``` ] .pull-right-text[ ### .header-multi[Plotting multiple datasets]  .plot-text[In this example we expand the pH reconstruction for the Round Loch of Glenhead and add a zonation using functions `addRPClust` to plot the dendrogram and `addRPClustZone` to plot zone lines on the figure. ]] --- class: inverse-multi .pull-left[ ### Adding multiple proxies .left-small[ ```r library(riojaPlot) library(readxl) library(dplyr) fpath <- system.file("extdata/allen1999.xlsx", package="riojaPlot") pollen <- read_excel(fpath, sheet="Pollen data", skip=2) pollen.chron <- pollen %>% select(1) pollen <- pollen %>% select(Pinus:`Other herbaceous taxa`) types <- data.frame(Name=colnames(pollen), Group="Woody taxa") types$Group[9:12] <- "Herbs" types$Group <- factor(types$Group, levels=c("Woody taxa", "Herbs")) mag <- read_excel(fpath, sheet="Magnetic susceptibility", skip=2) mag.chron <- mag %>% select(1) mag <- mag %>% select(`Mag Susc`) %>% mutate(`Mag Susc` = `Mag Susc` / 1000) loi <- read_excel(fpath, sheet="Loss on ignition", skip=2) loi.chron <- loi %>% select(1) loi <- loi %>% select("LOI (wt %)") BSi <- read_excel(fpath, sheet="Biogenic silica", skip=2) BSi.chron <- BSi %>% select(1) BSi <- BSi %>% select("BSi") rp1 <- riojaPlot(pollen, pollen.chron, groups=types, ymin = 0, ymax=102000, yinterval = 5000, yvar.name = "Age BP", scale.percent=TRUE, plot.groups=TRUE, do.clust = TRUE, plot.clust=TRUE, plot.zones = "auto", plot.cumul=TRUE, cex.cumul=0.6, srt.xlabel=45, plot.bar=FALSE, tcl=-0.1, cex.yaxis=0.7, cex.xlabel=0.7, cex.xaxis=0.5, xRight = 0.7, plot.line=FALSE ) rp2 <- riojaPlot(mag, mag.chron[, "Age BP", drop=FALSE], riojaPlot=rp1, xGap = 0.01, xRight=0.8, scale.minmax=FALSE, plot.bar=FALSE, plot.line=F, plot.symb=TRUE, symb.cex=0.2) rp3 <- riojaPlot(loi, loi.chron[, "Age BP", drop=FALSE], riojaPlot=rp2, xRight=0.9, scale.minmax=FALSE, plot.bar=F, plot.line=F, plot.symb=TRUE, symb.cex=0.2) riojaPlot(BSi, BSi.chron[, "Age BP", drop=FALSE], riojaPlot=rp3, xRight=0.99, scale.minmax=FALSE, plot.bar=FALSE, plot.line=FALSE, plot.symb=TRUE, symb.cex=0.2) ``` ]] .pull-right-text[ ### .header-multi[Plotting multiple datasets]  .plot-text[This example combines pollen, magnetic susceptibility, LOI and biogenic silica data recorded in a core from Lago Grande di Monticchio and published by Allen et al. (1999). The core spans the last c. 100 kyr and the four datasets are measured at different depth intervals and resolutions. The data are downloaded from the NOAA paleoclimatology data archive (https://www.ncei.noaa.gov/products/paleoclimatology). .reference-text[Allen, J.R.M. *et al*. (1999) Rapid environmental changes in southern Europe during the last glacial period. *Nature*, **400**, 740-743. ] ]] --- class: inverse-multi .pull-left[ ### Adding a lithology column ```r library(rioja) library(riojaPlot) library(purrr) poll <- aber$spec chron <- aber$ages names <- aber$names colnames(poll) <- names$Name max5 <- sapply(poll, max) > 5 max5_names <- names(max5[max5]) lithology <- data.frame( top= c(300, 325, 375, 400, 420, 440, 464, 510, 535), bottom=c(325, 375, 400, 420, 440, 464, 510, 535, 550), lithology=c("Peat", "Mud", "NS", "Mud", "Mud2", "SiltyMud", "SiltyMud2", "Mud3", "Silt"), colour=c("burlywood4", "saddlebrown", "NA", "brown4", "brown3", "navajowhite4", "navajowhite3", "brown4", "lightsteelblue") ) myfun <- function(x, style) { x %>% pwalk(~rect(0, ..1, 1, ..2, col=..4)) } rp <- riojaPlot(poll, chron, selVars=max5_names, lithology=lithology, sec.yvar.name="Age (years BP)", sec.ymin=6000, sec.ymax=14000, sec.yinterval=500, yvar.name="Depth (cm)", plot.sec.axis = TRUE, scale.percent=TRUE, fun.lithology=myfun) ``` ] .pull-right-text[ ### .header-multi[Plotting multiple datasets]  .plot-text[Adding a column showing lithogy is experimental but it is possible to show basic lithology with different colours and / or pattern fills. To do this create a data frame with at least 3 columns - the top and bottom of each lithological unit, and the fill colour for that unit. You also have a define a custom function that takes this data frame and draws the lithology. Here we create a function that uses apply to loop through each row in the lithology dataframe and plot a filled rectangle in the lithology column. Lithological units are taken from the original paper by Birks and Matthews (1978). .reference-text[Birks, HH & Mathews, RW (1978). Studies in the vegetational history of Scotland V. Late Devensian and early Flandrian macrofossil stratigraphy at Abernethy Forest, Invernessshire. *New Phytologist* **80**, 455-84. ]]] --- class: inverse-custom ## .footer-white[Custom functions] .pull-right[ ] --- class: inverse-custom .pull-left[ ### Customising the plot type for each variable ```r library(rioja) library(riojaPlot) poll <- aber$spec chron <- aber$ages names <- aber$names colnames(poll) <- names$Name # calculate of max of each column mx <- sapply(poll, max) #create a logical vector which is TRUE for taxa with max < 5 sel <- mx < 5 # define a custom function to plot symbols symb.fun <- function(x, y, i, nm, style) { sel <- x > 0 if (sum(sel) > 0) { points(rep(3, sum(sel)), y[sel], cex=0.3, pch=19, xpd=NA) } } # create a list of functions of length equal to the number of columns in the data funlist <- lapply(1:ncol(poll), function(x) symb.fun) # now set the elements of the list where we don't want to plot symbols to NULL funlist[!sel] <- list(NULL) # plot silhouettes and lines for taxa > 5% and apply our function to the others riojaPlot(poll, chron, scale.percent=TRUE, plot.poly=!sel, plot.bar = !sel, plot.line=FALSE, plot.exag=TRUE, cex.xlabel=0.6, fun.xfront=funlist) ``` ] .pull-right-text[ ### .header-custom[Custom functions]  .plot-text[This example uses a custom function to plots symbols instead of silhouettes for rare types in a pollen diagram. We first define a function that passes 5 variables from the main plot (x, y, i, nm, style - the x and y coordinates of each data point, the index number of the plot, the name of the variable being plotted, and the plot style, which is a list containing the various parameters controlling the diagram appearance). We then create a list of functions to be applied to be applied to each plot and set the elements of the list to `NULL` for those we don't want symbols plotted. We also turn off bars, lines etc. for the plots where we do want symbols. Finally, we pass our custom function to `fun.xfront`. `fun.xfront` calls the function after other plotting. A parallel argument `fun.xback` call a custom function to plot before other plotting. ]] --- class: inverse-custom .pull-left[ ### Enhancing the built-in plot types .left-small[ ```r library(riojaPlot) library(readxl) library(dplyr) fpath <- system.file("extdata/allen1999.xlsx", package="riojaPlot") pollen <- read_excel(fpath, sheet="Pollen data", skip=2) pollen.chron <- pollen %>% select(1) pollen <- pollen %>% select(Pinus:`Other herbaceous taxa`) types <- data.frame(Name=colnames(pollen), Group="Woody taxa") types$Group[9:12] <- "Herbs" types$Group <- factor(types$Group, levels=c("Woody taxa", "Herbs")) mag <- read_excel(fpath, sheet="Magnetic susceptibility", skip=2) mag.chron <- mag %>% select(1) mag <- mag %>% select(`Mag Susc`) %>% mutate(`Mag Susc` = `Mag Susc` / 1000) loi <- read_excel(fpath, sheet="Loss on ignition", skip=2) loi.chron <- loi %>% select(1) loi <- loi %>% select("LOI (wt %)") BSi <- read_excel(fpath, sheet="Biogenic silica", skip=2) BSi.chron <- BSi %>% select(1) BSi <- BSi %>% select("BSi") fun.gam <- function(x, y, i, nm, style) { tmp <- data.frame(x=y, y=x) gam <- mgcv::gam(y ~ s(x, k=50), data=tmp) x2 <- predict(gam, type="response") lines(x2, y, col="red", lwd=1) } rp1 <- riojaPlot(pollen, pollen.chron, groups=types, ymin = 0, ymax=102000, yinterval = 5000, yvar.name = "Age BP", scale.percent=TRUE, plot.groups=TRUE, do.clust = TRUE, plot.clust=TRUE, plot.zones = "auto", plot.cumul=TRUE, cex.cumul=0.6, srt.xlabel=45, plot.bar=FALSE, tcl=-0.1, cex.yaxis=0.7, cex.xlabel=0.7, cex.xaxis=0.5, xRight = 0.7, plot.line=FALSE) rp2 <- riojaPlot(mag, mag.chron[, "Age BP", drop=FALSE], riojaPlot=rp1, xGap = 0.01, xRight=0.8, scale.minmax=FALSE, plot.bar=FALSE, plot.line=F, plot.symb=TRUE, symb.cex=0.2, fun.xfront=fun.gam) rp3 <- riojaPlot(loi, loi.chron[, "Age BP", drop=FALSE], riojaPlot=rp2, xRight=0.9, scale.minmax=FALSE, plot.bar=F, plot.line=F, plot.symb=TRUE, symb.cex=0.2, fun.xfront=fun.gam) riojaPlot(BSi, BSi.chron[, "Age BP", drop=FALSE], riojaPlot=rp3, xRight=0.99, scale.minmax=FALSE, plot.bar=FALSE, plot.line=FALSE, plot.symb=TRUE, symb.cex=0.2, fun.xfront=fun.gam) ``` ]] .pull-right-text[ ### .header-custom[Custom functions]  .plot-text[This example reproduces the diagram from Lago Grande di Monticchio seen above but this time adds a gam smooth to the magnetic susceptibility, LOI and biogenic silica data. ]] --- class: inverse-custom .pull-left[ ### Customising axes and labels .left-small[ ```r library(readxl); library(dplyr) fpath <- system.file("extdata/LochChon.xlsx", package="riojaPlot") chon <- readxl::read_excel(fpath, sheet="Diatoms") sig_test <- readxl::read_excel(fpath, sheet="Sig_test") chon.diat <- chon %>% select(-Year) chon.year <- chon %>% select(Year) mx <- sapply(chon.diat, max) # # data has many rare species, remove these chon.diat <- chon.diat[, mx > 3] chon.groups <- data.frame(TaxonName=colnames(chon.diat)) %>% left_join(sig_test, by="TaxonName") %>% mutate(group = case_when(p_unadj < 0.1 & slope>0 ~ "Increasing", p_unadj < 0.1 & slope<0 ~ "Decreasing", TRUE ~ "Not sig"), group=factor(group, levels=c("Increasing", "Not sig", "Decreasing"))) %>% select(TaxonName, group) diat.order <- chon.groups %>% arrange(group) %>% pull(TaxonName) chon.ordered <- chon.diat %>% select(!!diat.order) names <- rep("", ncol(chon.ordered)) # create vector of empty names myfun <- function(x, y, i, nm, style) { usr <- par("usr") # extract the x and y data limits of the plot name <- bquote(italic(.(names(nm)))) # extract name from nm and italicise it text(usr[2], usr[4]+0.5, name, adj=c(0, 0.5), xpd=NA, srt=-90, cex=0.7) xval <- seq(0, usr[2], by=10) # create a vector of labels for the axis xlab <- rep("", length(xval)) axis(side=3, at=xval, labels=xlab, tcl=-0.2, cex.axis=0.5, mgp=c(3, 0.1, 0)) xlab <- as.character(xval); xlab[1] <- "" text(xlab, usr[4]-0.5, xval, cex=0.5, srt=-90, adj=c(1, 0), xpd=NA) # ad the x-axis vales } riojaPlot(chon.ordered, chon.year, groups=chon.groups, scale.percent=TRUE, yinterval=1, xlabels=names, # replace taxon names with a vector of blank names ylabel = " ", # suppress y-axis label plot.poly=TRUE, plot.line=FALSE, plot.groups=TRUE, cex.xlabel=0.6, cex.yaxis=0.5, labels.italicise=TRUE, cex.xaxis=0.5, col.group=c("darkgreen", "darkgrey", "darkred"), plot.bottom.axis=FALSE, min.width.pc=15, # set minimum size of x-axes xSpace=0.01, fun.xfront=myfun) ``` ]] .pull-right-text[ ### .header-custom[Custom functions] <img src="figures/custom_plot_3B-1.png", width="60%")/> .plot-text[ `riojaPlot` can also be used to plot multi-species ecological time series (or spatial transect) data. Here we plot diatom data from the UK Upland Waters Monitoring Network (https://uwmn.uk/). These are epilithic diatom samples collected annually from 1988-2018 from Loch Chon (Trossachs, Scotland). These data were collected to monitor the potential recovery of the loch from previous acidification. The file of diatom counts (expressed as relative abundance) also contains the results of a trend test based on a multivariate generalised linear model using the `manyglm` function in package `mvabund` used identify taxa that have a significant increasing or decreasing temporal trend. We first re-order the taxa according to their trend over time and plot the data with silhouettes, coloured to show taxa with significant increasing, decreasing or no trend. We then "virtually" rotate the figure so it can be viewed as a stacked diagram. To do this we modify the axis labels and position of the taxon names. We can't easily do this using `riojaPlot` styles so we can suppress the plotting of names and x-axes and create a custom function to plot elements in a more appropriate position. ]] --- class: inverse-custom .pull-left[ ### Customising the background ```r library(riojaPlot) library(rioja) # for Ponds data: see ?Ponds library(purrr) # reorder the rows in decreasing TP o <- order(Ponds$env$TP, decreasing=TRUE) ponds.diat <- Ponds$spec[o, ] ponds.TP <- data.frame(TP=round(Ponds$env$TP[o]), LakeName=Ponds$env$Name[o]) colnames(ponds.diat) <- Ponds$names$Name mx <- apply(ponds.diat, 2, max) ponds.sel <- colnames(ponds.diat)[mx > 10] cuts <- c(0, 100, 200, 500, 10000) scut <- which(diff(as.integer(cut(ponds.TP$TP, cuts))) != 0) + 0.5 scut <- bounds <- data.frame(upper=c(0.5, scut), lower=c(scut, 30.5), cols=c("orange", "red", "blue", "green")) fun.back <- function(usr, fig, style) { dim.col <- function(x, alpha) { # function to apply alpha value to a colour apply(col2rgb(x)/255, 2, function(x) rgb(x[1], x[2], x[3], alpha)) } style$user1 %>% pwalk(~rect(0.005, ..1, 0.995, ..2, col=dim.col(..3, 0.4), border=NA, xpd=NA)) } ylab <- expression(Total~Phosph.~(mu*gL^{-1})) riojaPlot(ponds.diat, ponds.TP, selVars=ponds.sel, yvar.name="LakeName", sec.yvar.name="TP", sec.ylabel=ylab, sec.yinterval=20, plot.sec.axis=TRUE, scale.percent=TRUE, plot.poly=FALSE, plot.line=FALSE, plot.bar=TRUE, col.bar="black", col.axis=NA, lwd.bar = 10, cex.xlabel=0.5, cex.yaxis=0.6, cex.xaxis=0.5, wa.order="bottomleft", labels.italicise=TRUE, las.xaxis=2, user1=bounds, fun.plotback=fun.back) ``` ] .pull-right-text[ ### .header-custom[Custom functions]  .plot-text[ Argument `fun.plotback` allows us to write a custom function for plotting in the background (ie. behind the plots for each variable). In this example we revisit the diatom surface sediment data from SE ponds. Previously we plotting these data with different coloured bars for four total phosphorus (TP) ranges (<100, 100-200, 200-500, >500) µg/L. Here we shade the background to show the TP ranges. First, we identify the sample positions of the breaks between the TP ranges, then create a data frame with the upper, lower limit of each range and an associated colour. In this example the y-axis is labelled with sample names, so numerically the axis runs from 1 to number of samples, so the y-axis bounds for each shaded rectangle is simply the sample number of boundary between groups plus 0.5). We then pass this data grame to argument `user1`. This data frame is passed into our custom plotting function as part of the `style` list. Finally, we pass out custom function to `fun.plot.back`. ]] --- class: inverse-intro, right, middle .pull-left[ ## Thanks for viewing! <br> <br>Steve Juggins, 2022 <br> Please send bug reports, comments and suggestions for improvement to [Stephen.Juggins@ncl.ac.uk](mailto:Stephen.Juggins@ncl.ac.uk) ]